MitoWorld had a few questions for Dr. Enríquez and his Lab
MitoWorld: The text notes that you enjoy thinking about “crazy” ideas. What is the “craziest” idea that intrigues you now?
One idea I’ve had for years is to replace the electron transport chain, complex I, III, and IV, for only two proteins. One would be an NADH dehydrogenase from yeast (for example NDi1), and the other would be an alternative oxidase (AOX). These can move electrons from NADH to oxygen, bypassing the traditional complex I/III/IV pathway. But here’s the crazy part: they don’t pump protons. That’s where proteorhodopsin comes in. It’s a light-driven proton pump. So, the idea is to express NDi1 and AOX in mitochondria to maintain redox balance and electron flux, and then express proteorhodopsin to regenerate the proton gradient using light. That proton gradient could then be used by ATP synthase to produce ATP.
But it gets even crazier. What if the AOX or NDi1 steps themselves could be engineered to emit light as a byproduct of electron transport? Then proteorhodopsin could be activated by that endogenous light, creating a feedback loop: electrons generate light, light drives proton pumping, protons drive ATP synthesis. In theory, with only three proteins—NDi1, AOX, and proteorhodopsin—we could reconstruct a functional, energy-producing electron transport system in mammalian mitochondria.
It’s very ambitious—perhaps impossible—but it has fascinated me since I was a student. The goal isn’t just synthetic biology, but to explore whether we can dissect the respiratory chain’s functions: electron flux, proton pumping, and ATP synthesis. These functions are often tightly coupled, but in biology, it’s not always beneficial for them to be. If we could separate them, we might gain insight into how they contribute independently to mitochondrial and cellular physiology. Whether I’ll ever be able to do it before the end of my career, who knows?
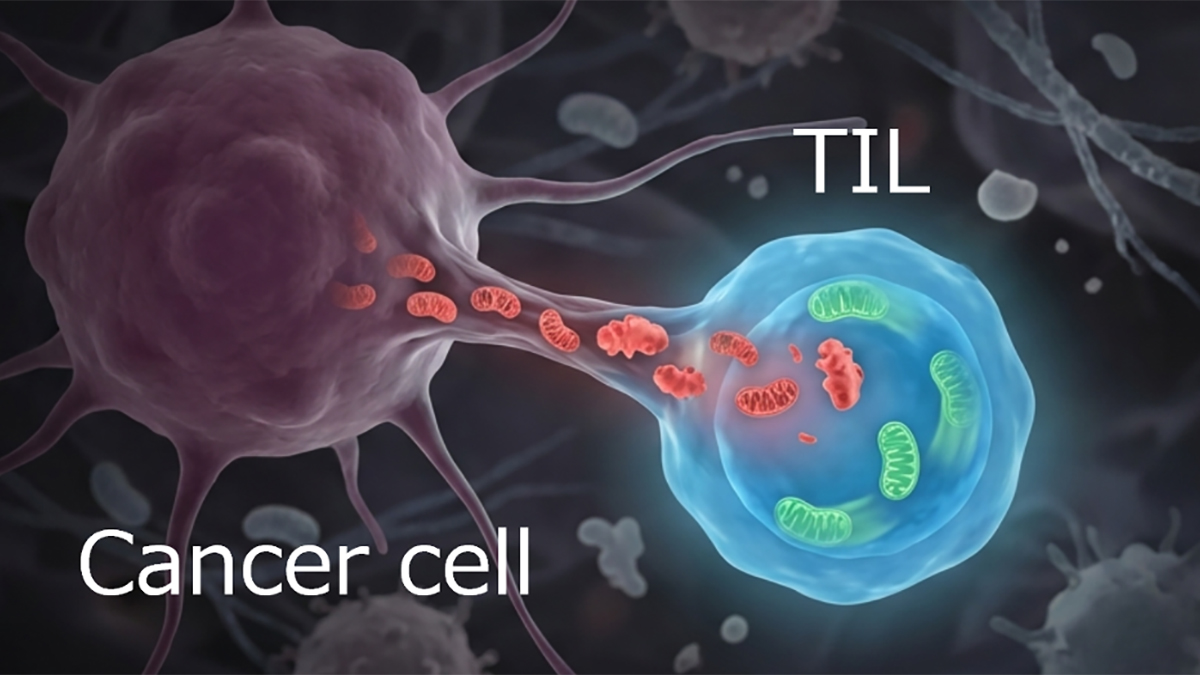
MitoWorld: A controversial hypothesis is that mitochondria move from cell to cell and that these transfers might serve as the basis for therapies. Do you have any thoughts about that possibility?
We not only have thoughts, but we also have experiments. In cardiac tissue, for example, mitochondria are packaged into membrane-bound structures we call “exopheres” and released. These are then taken up by neighbouring macrophages, which degrade them in a tightly regulated process. It’s a form of quality control but not necessarily a functional mitochondrial donation.
We’ve also shown, with Francisco Sánchez-Madrid, that mitochondrial components, especially mtDNA, can be transferred via immune synapses. The recipient cell sees that mtDNA as a danger signal, triggering antiviral responses.
Now, some studies—particularly in cancer—suggest that mitochondria can be taken up by recipient cells and remain functional, contributing to cell survival. However, while labeled mitochondria seem to transfer between cells, there’s little definitive evidence that they remain functional in the recipient cells or integrate into their networks.
The effects observed may often be immune related rather than metabolic. For instance, injecting mitochondria into blood has been proposed as a therapy, but the doses used are minuscule compared to endogenous mitochondrial populations in cells. Any physiological effects are more likely immune-mediated than due to actual mitochondrial “colonization.”
So, I think mitochondrial transfer happens, yes, but mainly as a signaling or quality control process. The notion of mitochondria behaving like autonomous, transferrable symbionts is still speculative.
MitoWorld: Can you speculate on how cells might regulate the specialization of mitochondria?
Between cell types, it’s mostly transcriptional regulation: different cells express different mitochondrial proteins or isoforms. But even within a single cell, mitochondria can be very different. One possible explanation is temporal regulation. A cell might express different sets of mitochondrial proteins at different times (e.g., day vs. night), and since mitochondria are relatively long-lived, this might create functionally distinct subpopulations. Another possibility is spatial regulation. Mitochondria traveling to different parts of the cell might encounter localized signals (e.g., specific mRNAs, post-translational modifiers, or regulators of protein import) that tune their function to local demands. In principle, if mitochondrial transfer between cells were robust, one could imagine specialized mitochondria being produced in one cell type and shared with another. But again, that’s still speculative.
MitoWorld: The role of mitochondria in energy production ensures that they are critical to cell and organismal health. Do you have any thoughts on how this might translate directly to health and aging?
Here I take a somewhat contrarian view. I don’t believe mitochondria drive aging. Aging is ultimately a consequence of entropy, a physical rather than strictly biological phenomenon. Biological systems resist entropy via repair and homeostasis, but over time, these processes lose efficiency.
That said, mitochondrial dysfunction is certainly part of the picture. As the cell accumulates damage (e.g., lysosomal inefficiency, metabolic imbalance, chronic inflammation), mitochondrial quality suffers. But so does everything else. For example, if oxygenation drops in the blood, the heart compensates by beating harder. Chronic compensation leads to hypertrophy and, eventually, pathology. The failure isn’t just in one organ or cell type; it’s in the integrated function of the whole.
So, while supporting mitochondrial function can help maintain cellular health, it won’t “cure” aging. In other words, mitochondria reflect the overall fitness of the cell. They are important, yes, but not supreme. Many age-related interventions that “target mitochondria” probably act through broader systemic effects: diet, exercise, metabolic control.
MitoWorld: Reversal of ATP synthase is counterintuitive. As you note, it might suggest a role in mitochondrial quality control. Can you elaborate on this hypothesis?
This might seem controversial at first: consuming ATP to pump protons. But it makes sense when you consider that the mitochondrial membrane potential must be tightly regulated. If it drops too low, apoptosis can be triggered. If it gets too high, ROS levels rise. So, reverse ATP synthase activity is a safety valve. It helps maintain an optimal membrane potential, even if doing so costs energy. In this light, ATP synthase becomes not just a producer of energy but a key regulator of mitochondrial homeostasis.
MitoWorld: FGR and OM1A are encoded by the nuclear DNA and imported into the mitochondria. That suggests a complex process with multiple potential therapeutic targets. Do you have thoughts about the most effective mechanism to inhibit it if it is not simply the enzymatic activity?
The challenge is that these enzymes have multiple substrates. Inhibiting their entire activity entirely can have off-target effects. A better strategy is to selectively block their interaction with specific targets, while leaving other interactions intact. Now that we know their structures, we can start designing molecules that interfere with specific protein-protein interactions to disrupt pathological effects while preserving beneficial ones. This is a more nuanced and promising strategy for targeting multifunctional mitochondrial regulators.
MitoWorld: One of the key questions in mitochondrial biology is how the mitochondria and the nucleus work together to keep things running smoothly—or not—when something goes wrong. Do you think your population studies might help us understand that relationship better?
This is a very important question. One of the biggest challenges in mitochondrial biology is that most mitochondrial protein complexes are assembled from proteins encoded by two different genomes: the mitochondrial genome and the nuclear genome. This creates a need for very tight coordination between the two, despite the fact that they evolve at different rates and under different selective pressures.
From a population genetics perspective, we can use large datasets to ask: (I) Which positions in these proteins are highly conserved (i.e., intolerant to change)? (II) Which positions vary more across individuals or species (i.e., more permissive to variation)? Some variants in mtDNA are never observed in the population, suggesting they are incompatible with nuclear-encoded partners.
We recently created a database scoring every amino acid in mitochondrial and nuclear-encoded ETC proteins for its permissivity—how often it varies across species and individuals. This helps us identify evolutionarily constrained sites where mitonuclear compatibility is critical.
We also see that mitochondrial and nuclear subunits in physical contact tend to co-evolve, whereas regions not in contact evolve more independently. This helps us map critical interaction interfaces and might uncover regulatory hotspots we don’t yet understand.
So yes—population studies can teach us a lot about mitonuclear compatibility, evolutionary constraints, and potentially, disease susceptibility.