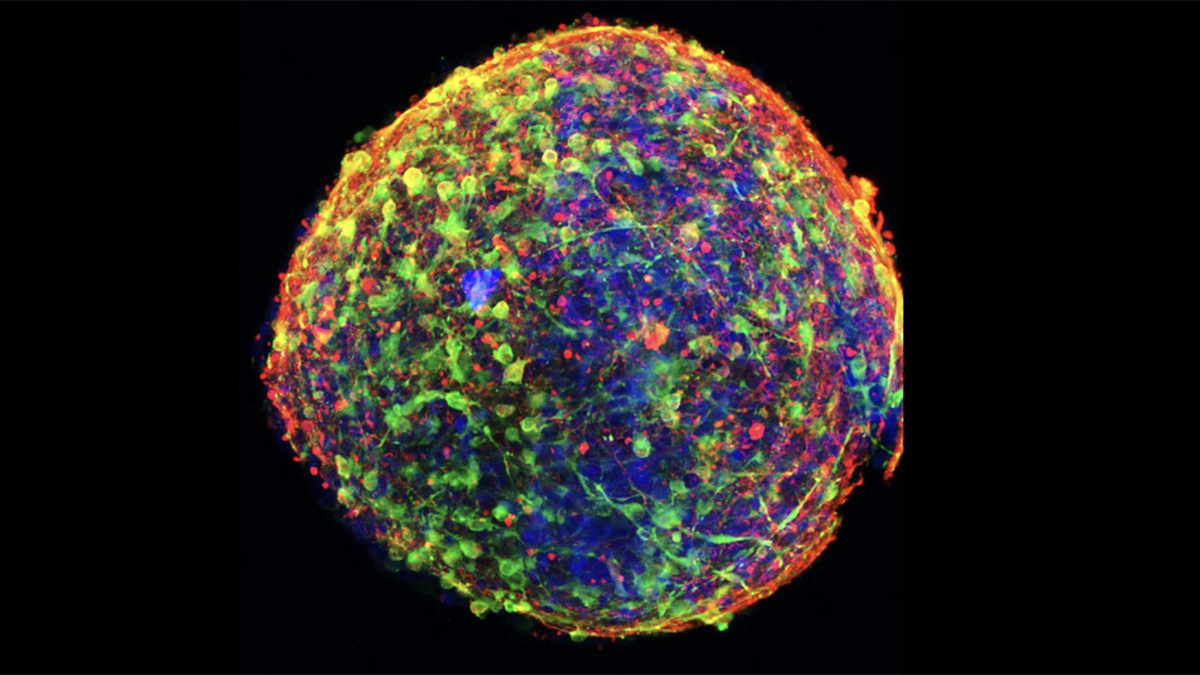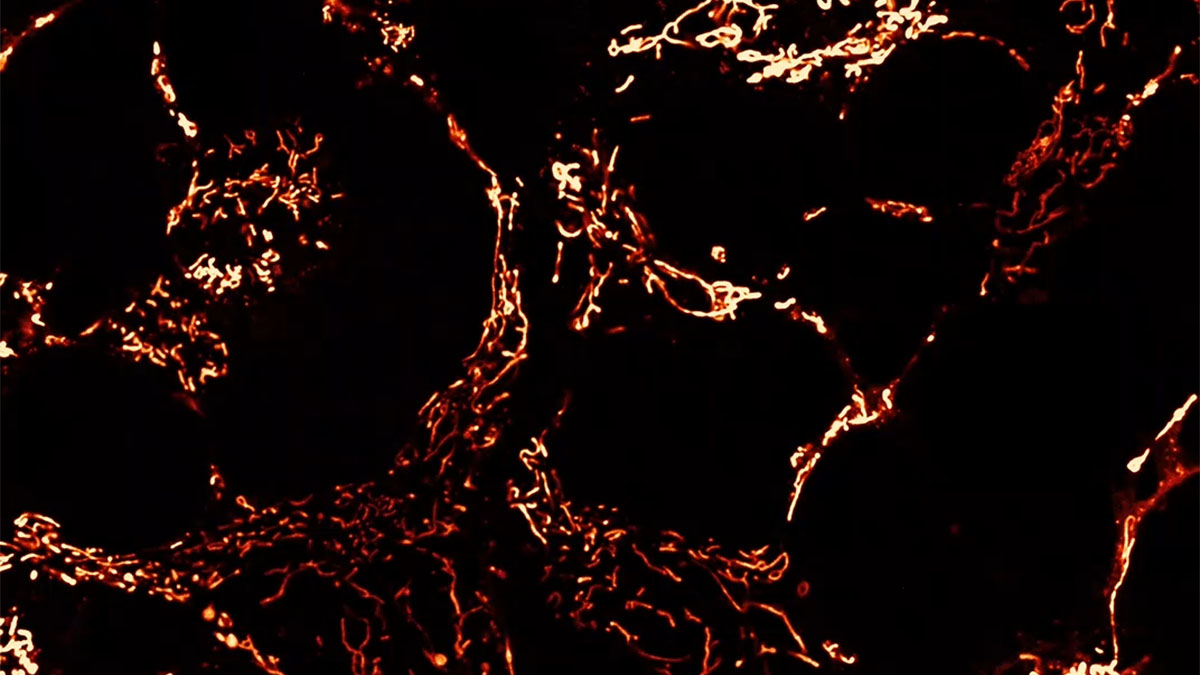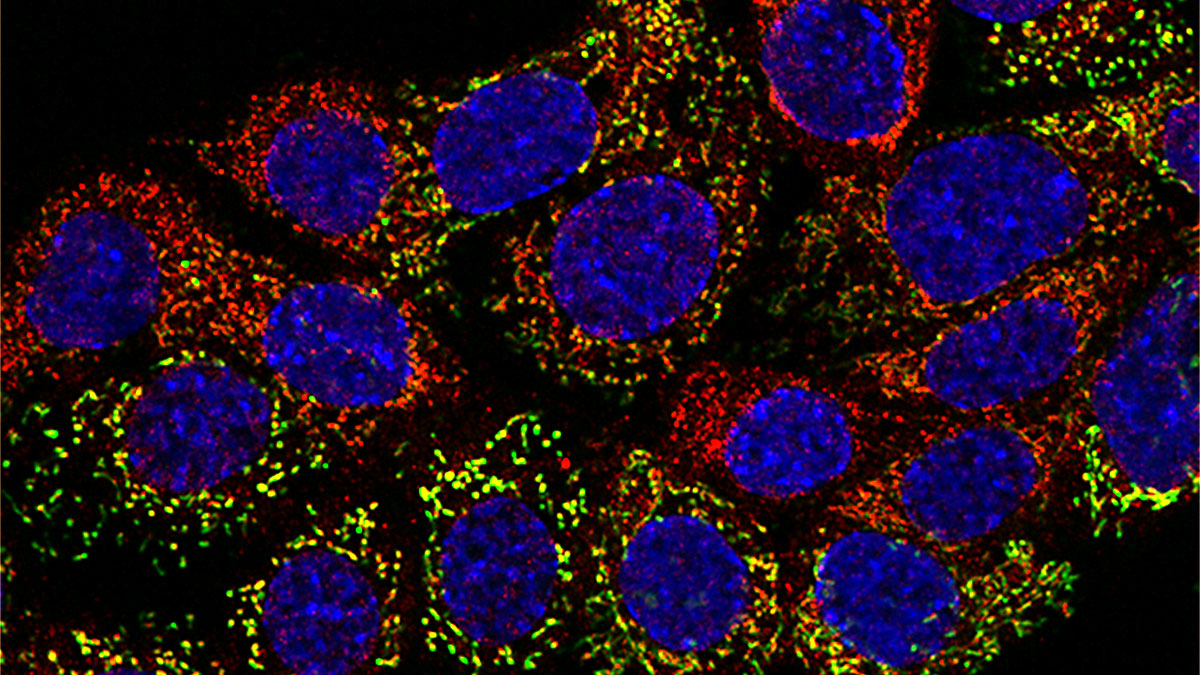
8-oxo-guanine (8OHdG, red) mitochondrial staining (green), and merge with DAPI for nuclei (blue) in mouse tubular epithelial cells under acute oxidative stress (H2O2 for 30 min).
Mitochondrial DNA Mutations and Aging
In a paper published in Science,1 Samir M. Parikh and an inter-institute research team explored the relationships among mitochondrial DNA (mtDNA) mutations, acute injuries, and chronic damage to the kidney. They found that the injuries result in mutations that hinder the mitochondria’s key activities and leave the organ more susceptible to future damage. Importantly, they found that this damage was amenable to metabolic treatment.
Mutations to mtDNA have been implicated as a driver of aging. The mitochondrial genome is much smaller than the nuclear genome. However, each cell contains hundreds to a thousand or more mitochondria, and each mitochondrion contains two to 10 copies of the mtDNA. Interestingly, mutations to mtDNA accumulate with aging, and that accumulation accelerates after age 70. The mutations to some but not all copies of the mtDNA in each mitochondrion result in differences in those genomes. This is called heteroplasmy.
The team used a mouse model for acute and chronic injuries to kidneys to examine the effects of mtDNA mutations. They found that the mutations accumulate and that they reduce gene expression, oxidative metabolism, and resistance to oxidative injury. Exogenous supplies of adenosine, but not other nucleosides, improved the function of the injured kidneys. Further studies identified loss of the key enzyme adenylate kinase 4, encoded in the nuclear genome, as the reason for the loss of ATP and the ability of adenosine to improve outcomes.
When they examined a large human cohort of 370,000 individuals from the UK biobank, they found that mtDNA mutations represented a risk factor for future acute injury and associated with the severity of chronic kidney disease. These results confirmed the association between mtDNA mutation burden and kidney injury.
These findings also confirm the kidney as an excellent model for the study of aging. Finally, they suggest that injuries to other organs, such as stroke and myocardial infarction, might be influenced by the effects of accumulating mtDNA mutations.
Conversation with Dr. Parikh and first author Dr. Huihui Haung
MitoWorld: What is the next likely goal in your research to follow up on this work?
SMP and HH: There are so many different directions to take this work, and they’re all interesting! On the translational side, does the relationship between mtDNA mutation burden and kidney disease hold in other human cohorts? More fundamentally, what mechanisms does a cell employ to monitor the fidelity of its mtDNA? Is it just overall oxidative function, or something more specific? Do cells in other organs composed of long-lived cells endowed with a high mitochondrial content also exhibit time and injury-dependent mtDNA mutations?
MitoWorld: There are so many copies of the mtDNA in each cell that it seems hard to understand how mutations to some of them can cause trouble. Do you have any speculation on what the threshold is for those mutations to be a problem?
SMP and HH: There are a few possibilities. First, the heteroplasmic single-nucleotide variants that are a focus of the manuscript represent one kind of mtDNA damage. Other assays largely relegated to the supplementary materials accompanying the paper demonstrate the rapid appearance and persistence of more profound changes to the mtDNA, such as indels. Second, we found that artificially introducing heteroplasmic mtDNA mutations by compromising the 3-5′ exonuclease activity of POLG sufficed to reduce oxidative function in renal tubular epithelial cells. Whether these changes affect tRNA function or subunits of the electron transport chain, or both, remains to be determined. Finally, in the cellular and mouse model data, we analyzed heteroplasmy without a threshold, but because the human data have so many orders of magnitude higher replicates per group, we were able to employ a more stringent variant allele frequency cutoff of 5%.
MitoWorld: Interestingly, the damage in your model was to a nuclear gene. How do you see the mtDNA mutations related to that enzyme?
SMP and HH: We do not yet have direct evidence that the enzyme AK4 regulates mtDNA mutations. However, we know that communication between the mitochondrial and nuclear genomes is essential to maintain mtDNA heteroplasmy within a safe threshold. AK4 is a nuclear-encoded mitochondrial protein and a key enzyme that regulates cellular ATP levels. Its expression is highly responsive to mitochondrial DNA damage. We speculate that AK4 may serve as a bridge or sensor that transmits deleterious mitochondrial signals to the nucleus, coordinating energy balance and mitochondrial fate. Further studies are needed to test this hypothesis.
MitoWorld: Damaged mitochondria have been implicated in aging and your current work also suggests that. What mechanisms might be involved? Is it just the slow deterioration of energy production or another mechanism?
SMP and HH: This is a really interesting question. Recent exciting work also conducted in the UK Biobank resource suggests that the nucleus has an important role in determining the burden of mtDNA mutations over a lifetime.2 The nuclear genes they implicated through a GWAS analysis were either the same or highly related to the ones we arrived at experimentally. The notion of a cell’s battery running down with age is intuitive and appealing. It is also possible that there is gain of toxic function as the mitochondrial code degrades. More work is needed to dissect among these possibilities.
MitoWorld: Can you envision how reducing mtDNA heteroplasmy might slow the processes of aging?
SMP and HH: Restoring the mitochondrial code may itself improve the oxidative function of the cell’s organelle. One could also imagine that the cellular processes involved in balancing the cell’s complement of mitochondria—namely the balance between mitochondrial biogenesis and mitochondrial clearance through mitophagy and the lysosome—wind up having multiple beneficial effects on overall cellular function.
MitoWorld: What about mitochondria interested you in the first place?
SMP and HH: As your readers appreciate, mitochondria are weird in many wonderful ways. Their membranes are composed of different lipids, their proteins start with a uniquely modified N-terminal amino acid, and the mtDNA genome lacks introns and the code to generate the vast majority of proteins that make up mitochondria, but exists in variable and polyploid amounts. The generation and dissolution of mitochondrial mass can be uncoupled entirely from cellular replication, and even the physical structure and networked appearance of mitochondria within cells are highly dynamic. Our bodies’ reliance on these weird organelles is clear from genetic experiments of nature.
The human data on monogenic mitochondrial syndromes are clear that kidney health is frequently impaired. It’s very easy to visualize mechanical work, such as skeletal or cardiac muscle contraction, requiring lots of ATP. But the tubular epithelium in the kidney is doing an enormous amount of chemical work. It might surprise the readers to know that, in humans, this specialized epithelium is responsible for the active transport of more than one pound of sodium back into the body every single day, atom-by-atom, against powerful electrochemical gradients. That’s more than 10e25 cations being pushed “uphill” back into the body from the crude filtrate every day. This apparatus is so exquisitely tuned that even a reduction to 10e24 sodium atoms would be lethal in a matter of minutes.
References
1 Huang H, Wang Y, Zsengeller ZK, Gorham JM, Vemireddy V, Clark AJ, Pan H, Dreyfuss JM, Jotwani V, Shlipak MG, Sarnak MJ, Parikh CR, Thiessen-Philbrook H, Katz R, Waikar SS, Lake NJ, Lek M, Shi W, Puiu D, Hong YS, Seidman JG, Arking DE, Parikh SM (2025) Reversible compromise of physiological resilience by accumulation of heteroplasmic mtDNA mutations. Science 390: 164–172.
2 Gupta R, Kanai M, Durham TJ, Tsuo K, McCoy JG, Kotrys AV, Zhou W, Chinnery PF, Karczewski KJ, Calvo SE, Neale BM (2023) Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 620: 839–848.