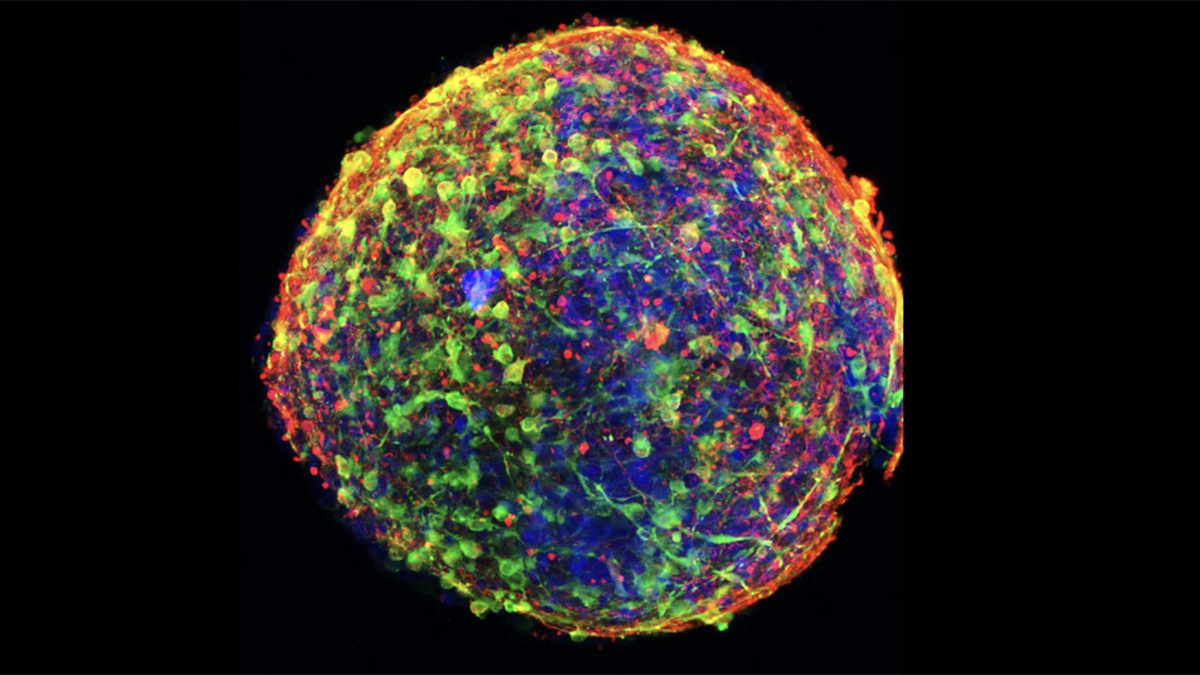
Macrophages from the Polg R292C mouse model of MtD undergo rapid inflammatory cell death after infection with Pseudomonas aeruginosa.*
Innate Immune System Dysfunction in Mitochondrial Diseases
In a paper in Nature Communications, a multi-institution research team, led by Phillip West at The Jackson Laboratory, describes hyperactivity of the innate immune system in models of polymerase gamma (PolG)-related mitochondrial disease (VanPortfliet et al., 2025). This work advances understanding of how mitochondrial diseases impact the immune system and identifies potential therapeutic targets to limit immunopathology and other infection-associated complications.
Mitochondrial diseases (MtDs) are the most common inborn errors of metabolism. Although patients with MtD do not appear to have more viral and bacterial infections than others, emerging research suggests infections can result in more severe outcomes, including sepsis and death. The relationship of MtDs and inflammation has therefore become a topic of considerable interest in the research community. Mitochondrial dysfunction can activate the innate immune system, which responds with inflammation that, when unregulated, further damages mitochondrial activity.
In their paper, West’s team delved further into this problem. More specifically, they examined two mouse models that carry deleterious mutations in the PolG gene (PolgD257A and PolgR292C). They found that these mutations, which impact mitochondrial DNA (mtDNA) stability, result in chronic activation of the type I interferon (IFN-I) pathway in immune cells and tissues. Furthermore, they uncovered that IFN-I hyperactivates another immune sensor called caspase-11, which senses bacterial cell wall components and promotes inflammatory cell death in macrophages. This form of cell death, called pyroptosis, is critical for control of bacterial infections, but must be tightly regulated because it promotes the release of cytokines and other factors that lead to a strong inflammatory response. When innate immune cells from the PolG mutant mice were infected with bacteria, they underwent pyroptosis much more readily and caused a dramatic increase in inflammatory responses. This overactive innate immune response was also seen when PolG mutant mice were infected with bacteria.
Although these PolG mutant mice do not recapitulate all aspects of PolG-related MtDs, chronic activation of the innate immune system, increased inflammatory responses, and other symptoms are seen in MtDs in humans. Thus, this experimental system is an excellent model for studying innate immunity in MtDs.
A Conversation with Dr. West
MitoWorld: What caused you to become interested in mitochondria and MtDs?
West: I have been studying the interplay between mitochondria and the innate immune system since my PhD training at Yale. I somewhat stumbled into mitochondrial biology during my thesis research, but have been fascinated by these organelles ever since. As a postdoctoral fellow with Gerry Shadel, I found that mtDNA release is a potent trigger of interferon and inflammatory responses. As all of our early work was in cells, I wanted to translate our findings into animal models when I opened my own lab. We hypothesized that because MtDs have dysfunctional mitochondria and often exhibit mtDNA instability, there may be an unappreciated role for immune dysfunction in these diseases. We are addressing this hypothesis in mouse models of MtD, including the PolG mutants used in this paper, but are also striving to translate our results into understanding immune dysfunction in human MtDs.
MitoWorld: Under normal circumstances, the immune system is carefully regulated. Too little control is thought to allow cancers to grow. Too much results in autoimmune diseases. MtDs are yet another source of immune dysregulation. Do you have ideas about how to follow up your work in humans?
West: We are working collaboratively with Dr. Peter McGuire’s group at the NIH/NHGRI, who are also studying in immune dysregulation in MtDs. We were fortunate to be included on Peter’s recent study (Warren et al., 2023) that revealed interferon and inflammatory gene signatures in the white blood cells of patients with diverse MtDs. There was significant overlap in the immune signatures seen in patient cells and two of our mitochondria mutant mice, so we do feel our animal studies correlate with human data. Our goal now is to identify immunotherapeutics that may be used to restore proper immune function and limit infection-related complications in individuals with MtDs.
MitoWorld: It’s interesting that MtD patients are more susceptible to infections and have an enhanced innate immune response. During the Covid pandemic, any vaccination was thought to activate the innate immune system and protect (to a degree) against coronavirus infection. Is the MtD case, just another example of the immune system gone awry?
West: This is an interesting question. I think it is important to highlight that the immune phenotypes in MtDs will probably be diverse and not manifest in exactly the same ways. For example, those with Barth syndrome often have neutropenia, or to few neutrophils, and are susceptible to bacterial infections. In addition, Dr. Anu Suomalainen-Wartiovaara’s group recently reported reduced antiviral responses in patient samples and mice carrying the PolG MIRAS allele, suggesting that there may be dramatic differences in immune phenotypes even within PolG-related MtDs (Kang et al., 2024). Other MtDs may cause hyperactive innate immunity, whereas some may lead to problems with adaptive immunity (i.e., antibodies and T cells). We are early in these studies, and MtDs are rare diseases, making it often difficult to obtain large patient cohorts for study. However, we can rapidly advance the field by generating new, more relevant animal models of MtD and coupling these findings with data from human studies.
MitoWorld: MtDs manifest at different ages. Do you have any ideas about what might activate the immune system in an MtD?
West: We hypothesize that mitochondrial dysfunction in MtDs basally alters the tone of immune cells. This is likely due to small amounts of cytokines and other stimulatory factors being released constitutively. For example, we showed that the aberrant release of mtDNA and other nucleic acids triggers the innate immune system in the absence of infection. Metabolic alterations in MtDs can also profoundly impact immune cell development and function. In the context of infection, innate immune cells, such as macrophages, may mount an overactive response, and this can feed forward to damage mitochondria and trigger subsequent rounds of mtDNA release or elevate metabolic crisis.
MitoWorld: So many of the former mitochondrial genes are now part of the host genome. Could mutations in those genes cause similar problems in mitochondria?
West: Most of my lab’s work has focused on examining innate immune responses in mouse models where nuclear-encoded mitochondrial genes are missing or mutated. However, others are examining immune responses in patients and animal models with particular disease-relevant mtDNA mutations. For example, Dr. Martin Picard has shown that cells from patients carrying a single, large-scale mtDNA deletion have blunted inflammatory cytokine responses (Karen et al., 2022). In contrast, a mouse model carrying a heteroplasmic mtDNA mutation (m.5019A>G) mirroring that seen in humans exhibit a hyperinflammatory immune status characterized by elevated interferon (Marques et al., 2025). Therefore, mutations in nuclear and mtDNA encoded mitochondrial genes can impact the immune system.
MitoWorld: How do you plan to extend this research?
West: My lab and colleagues at JAX are working to expand the toolkit of mouse models for MtDs, and we are excited to send our new models into labs around the globe. I am quite hopeful that MitoWorld, the UMDF, the PolG Foundation, and other advocacy groups will better unite researchers examining immunological issues in animal models and patients with MtDs.
* Two hours after infection, macrophages were stained with antibodies and dyes to mark the cell membrane (white), mitochondria (green), the nucleus (blue), and Pseudomonas bacteria (magenta). Cells were then imaged on a confocal microscope. The macrophage at the bottom right is undergoing pyroptosis, an inflammatory cell death pathway resulting in nuclear condensation, membrane permeabilization, loss of mitochondria, and release of cytokines.
References
Kang Y, Hepojoki J, Sartori Maldonado R et al. (2024) Ancestral allele of DNA polymerase gamma modifies antiviral tolerance. Nature 628: 844–853.
Karan KR, Trumpff C, Cross M, Engelstad KM, Marsland AL, McGuire PJ, Hirano M, Picard M (2022) Leukocyte cytokine responses in adult patients with mitochondrial DNA defects. J Mol Med (Berl) 100: 963–971.
https://pmc.ncbi.nlm.nih.gov/articles/PMC9885136/ (PubMed Central)
https://link.springer.com/article/10.1007/s00109-022-02206-2 (behind paywall)
Marques E, Burr SP, Casey AM, Stopforth RJ, Yu CS, Turner K, Wolf DM, Dilucca M, Tyrrell VJ, Kramer R, Kanse YM. An inherited mtDNA mutation remodels inflammatory cytokine responses in macrophages and in vivo. bioRxiv 2025 Jan 5:2025-01.
https://www.biorxiv.org/content/10.1101/2025.01.05.631298v1
VanPortfliet JJ, Lei Y, Ramanathan M, Guerra Martinez C, Wong J, Stodola TJ, Hoffmann BR, Pflug K, Sitcheran R, Kneeland SC, Murray SA, McGuire PJ, Cannon CL, West AP (2025) Caspase-11 drives macrophage hyperinflammation in models of Polg-related mitochondrial disease. Nat Commun 16: 4640.
https://doi.org/10.1038/s41467-025-59907-8
Warren EB, Gordon-Lipkin EM, Cheung F et al. (2023) Inflammatory and interferon gene expression signatures in patients with mitochondrial disease. J Transl Med 21: 331. https://doi.org/10.1186/s12967-023-04180-w